Hyperparameter Space






The world is the enzymes' playground and we are but their vessels. We wage their wars, we nurture them, we help them evolve, grow, and replicate. Life really is all about them. Fortunately, we’ve also learned to harness them for our ends, using them to synthesize and edit DNA, digest harmful materials, and, crucially, produce a vast array of molecules with a plethora of applications ranging from agriculture to pharmaceuticals. Compared to what organic synthesis techniques can accomplish with long sequences of basic chemical modifications, enzymes can achieve more complex molecular structures with less steps, at enviable ambient temperatures, many times more yield, and a higher chance of bioactivity. It is no wonder that the majority of drugs in the market have been natural products, which result from millions of years of enzymes evolving intertwined in biosynthetic pathways. And because of these reasons, there have been several efforts to transplant, evolve, combine, and engineer the enzymes and their biosynthetic pathways to produce known and novel chemical matter. This is the field of biocatalytic retrosynthesis. As of 2019, and really as of today, by far the best review of this area that I could find is by Lin, Warden-Rorthman, and Voigt – it’s very comprehensive, has a beautiful list enumerating biocatalytic retrosynthesis pathways that have been successfully executed and its references comes out at a whopping 352 papers. Just for a moment, feast your eyes on the pathways in these images, from the Lin, et al review:
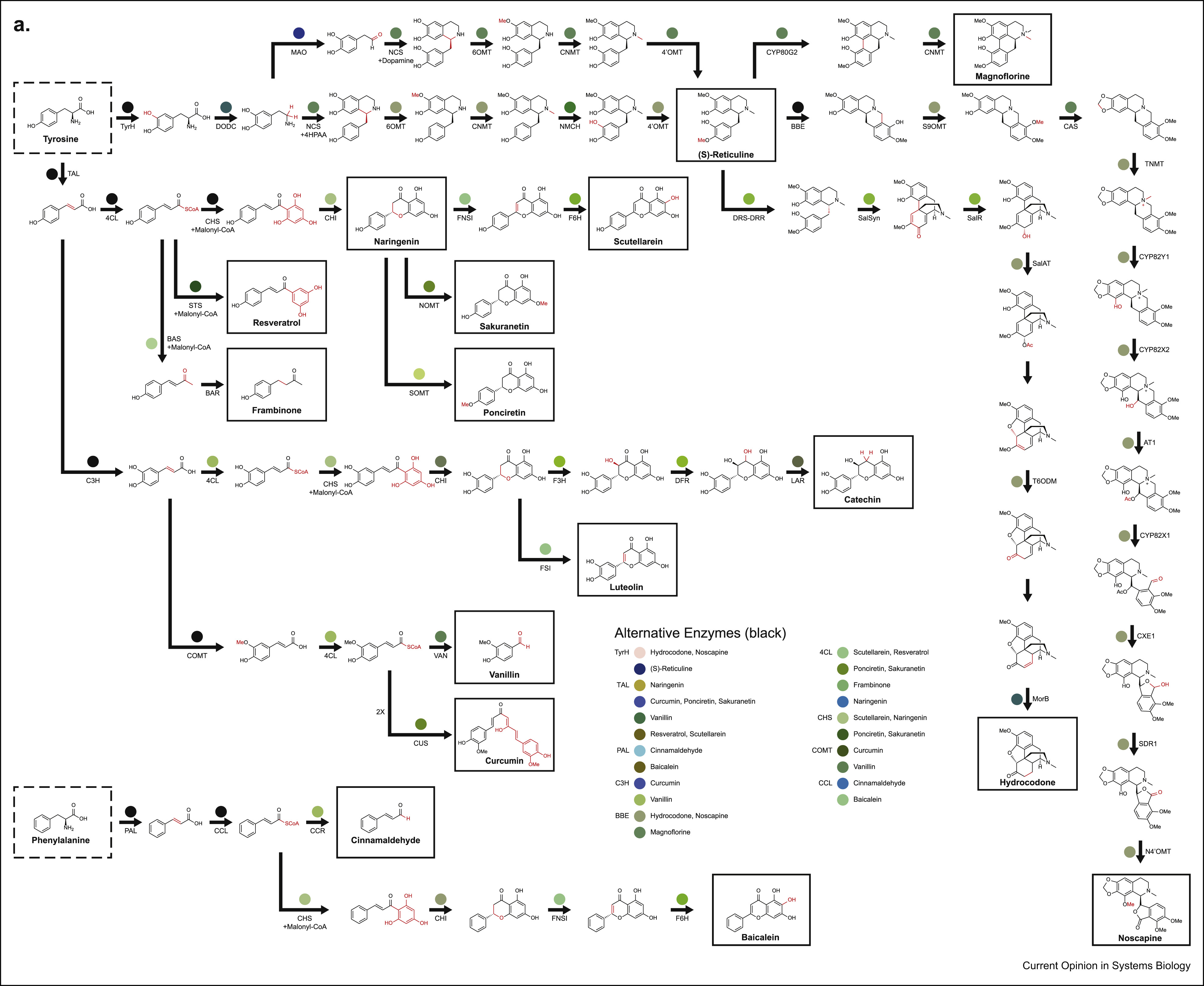 From Warden-Rorthman and Voigt, notice noscapine on the right
From Warden-Rorthman and Voigt, notice noscapine on the right
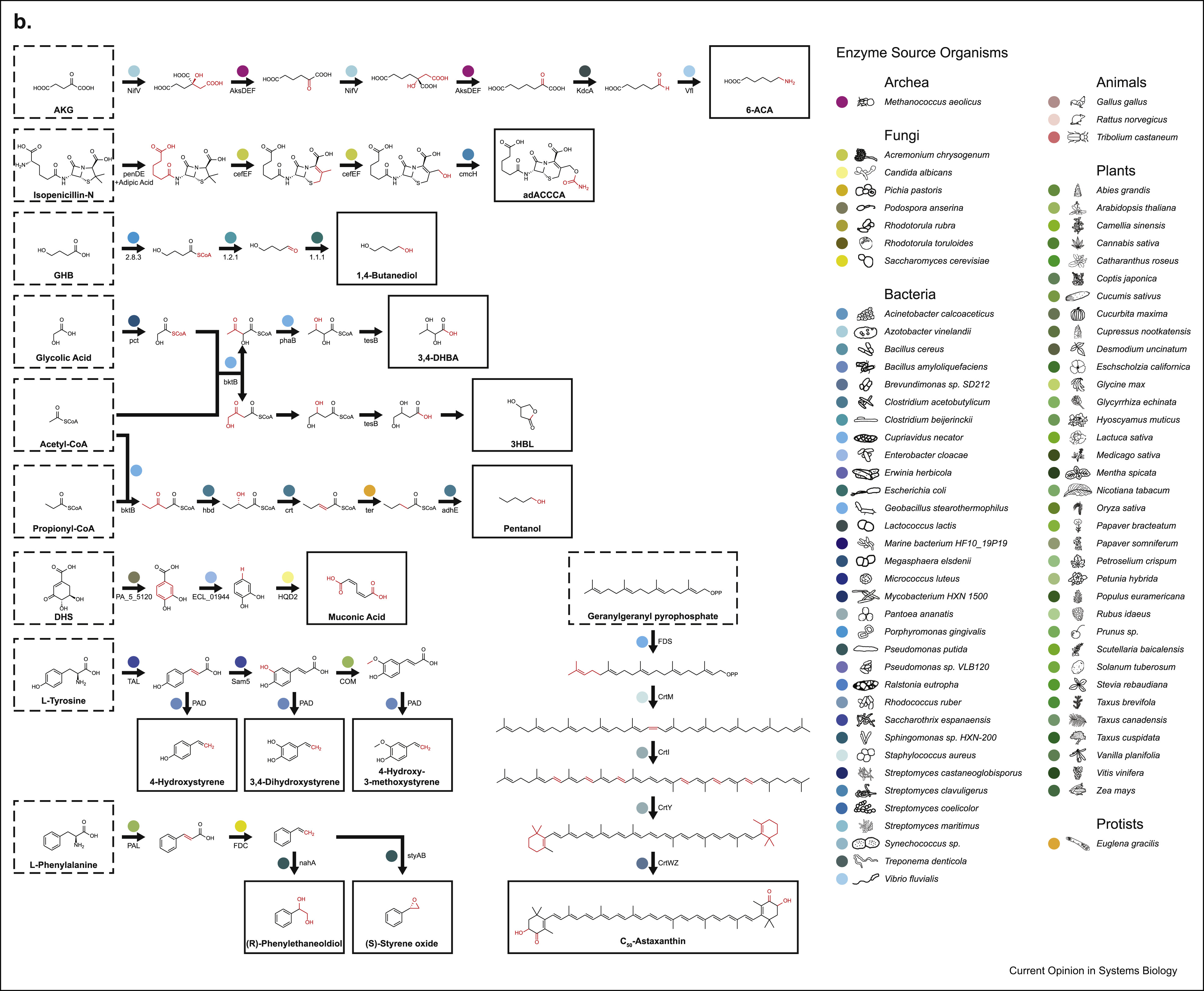 We’ll also talk a little bit about (S)-styrene oxide down-left
We’ll also talk a little bit about (S)-styrene oxide down-left
What’s striking is the diversity of molecules, how big they are, and the diversity of organisms they span. Just look at noscapine, on the first image to the right. Noscapine is an alkaloid anticancer drug and the starting point for various important drugs called “noscapinoids”. It is naturally produced by the opium poppy, a plant that uses 10 enzymes to make it. Now, one can of course just cultivate and harvest lots and lots of poppies to get noscapine, but this is terribly inefficient and probably won’t get you the amounts of product you need for downstream applications. But we know the 10 genes that make it, right? Couldn’t we just transplant the pathway (formally known as heterologous expression) into, say, yeast and culture vats of noscapine-producing beer fermentors? For some natural products, you can actually do this out of the box, but for noscapine, you need a lot more ingredients that what is already available in yeast. So you have to engineer your yeast to be able to apply the noscapine gene cluster transformation to actually get noscapine. Turns out, as the figure suggest, you need 8 more enzymes, many from different organisms, to actually make this work (and this is after optimization, the first attempt that got this to work needed 25 enzymes in total). The Smolke lab did this and this effort eventually led to their current enterprise in Antheia Bio. This is the essential task of biocatalytic retrosynthesis: starting with a cluster of genes known to produce a compound, selecting a host where to perform heterologous expression (which could be a cell free system too), then finding all other enzymes needed to produce the raw material for the pathway. The farther away your host is from the organism producing the compound and the more specific the compound is to that organism the harder it will be to achieve its biocatalytic retrosynthesis. The cool thing about succeeding though, is that once you can do biocatalytic retrosynthesis of a compound in an easy to grow host, the rest is host and enzyme optimization to produce enough titers.
Expanding on the natural compounds through a biocatalytic network
Though biocatalytic retrosynthesis of known natural products can get difficult even when you know the original pathway that produces the compound, trying to retrosynthesize something that doesn’t occur in nature at all presents an order of magnitude more of a challenge and there are only a handful of examples of getting this to work. Even then, the chemicals synthesized are typically not far off from something found in nature and are mostly one or two-step transformations. An example of this is (S)‐styrene oxide (see the second image above, down and to the left) that is used as a precursor for several biocides and that can be obtained by applying a styrene monooxygenase to styrene, which can be obtained from trans-Cinnamate that in turn is produced via a phenylalanine via a ferrulic acid decarboxylase. While the final steps in producing (S)-styrene oxide require only a couple of enzymes, these last steps needs to be properly set up in the host via an additional set of reactions. This is a common theme in biocatalytic retrosynthesis, where hosts are engineered and augmented with heterologous pathways that can be used as stepping stones for synthesizing additional compounds. Every successful implementation of a pathway both builds on and contributes to a large network of enzymatic reactions that serve as a catalog for future retrosynthesis efforts. This curation effort sounds like a lot of manual work, so what if we wanted to automatically map an arbitrary compound to a biocatalytic pathway that produces them? One may be tempted at first to conceptualize biocatalytic retrosynthesis as a network where nodes are reactants and edges are reactions. This can be, however, only an approximation since the number of reactants can be huge (and the largest of them are proprietary, e.g. with Reaxys listing more than 35 million chemicals) and each reaction does not only encode a relationship between two chemicals, but is rather an instantiation of much more general rules that also apply to other chemicals, generating more and more reactants. Thus, enumerating all reactants and reactions possible becomes unfeasible and one is faced with two combinatorial challenges: how to efficiently represent reaction rules and how to explore the chemical space generated by such rules. Further, biocatalytic retrosynthesis adds the challenge of incorporating the information contained in enzymatic sequences to the reactions they might catalyze. Let’s examine these challenges in more detail.
Challenge 1: Representation and expansion of reaction rules
Reaction rules are functions between reactants and a product that are ideally invertible. Building such functions from available reaction databases can be done in a number of ways. The simplest is to choose molecular representations or signatures for the reactants, projecting them in some fingerprint space, and representing the reaction as the difference between sums of products and sums of reactants, weighed by their stoichiometry coefficients. If the molecular representations are robust, the reaction signatures will be unique and the signatures of similar reactions will be closer together in signature space, providing additional information that can be used to group and/or infer new reactions. In this same manner, common structured molecular representations like SMILES and SMARTS have been adapted to represent reactions as well. Another strategy involves aligning products and reactants and encoding such an alignment in a fingerprint.
Many times, these rules are derived from a database that is expected to be incomplete as many enzymes tend to catalyze in generic patterns. This requires abstracting or expanding the reactions in such a way that best describes what we think an enzyme is doing (e.g. “add some moiety to any ketone”). Abstracting these rules – effectively learning a general pattern from many existing reactions – requires identifying common reaction “cores” in the substrates that are transformed by the enzyme into the products. Reaction core identification is identical to the maximum common substructure problem, which fortunately can be approximated fairly well in small molecules as the graphs tend to be small, and probably because of that I’ve only seen one paper that uses modern ML techniques that tackle this problem – you don’t really need to use cutting edge techniques. In addition to proper rule abstraction, one can further expand the rules derived from a reaction database by learning the space of rule-enzyme pairs, like this paper does using Gaussian processes. This last step allows assigning a likelihood to an arbitrary enzyme-reaction pair. That way, not all reaction rules used need to be annotated with an enzyme, but rather the rules can be assigned an enzyme post-facto in the restrosynthesis pathway.
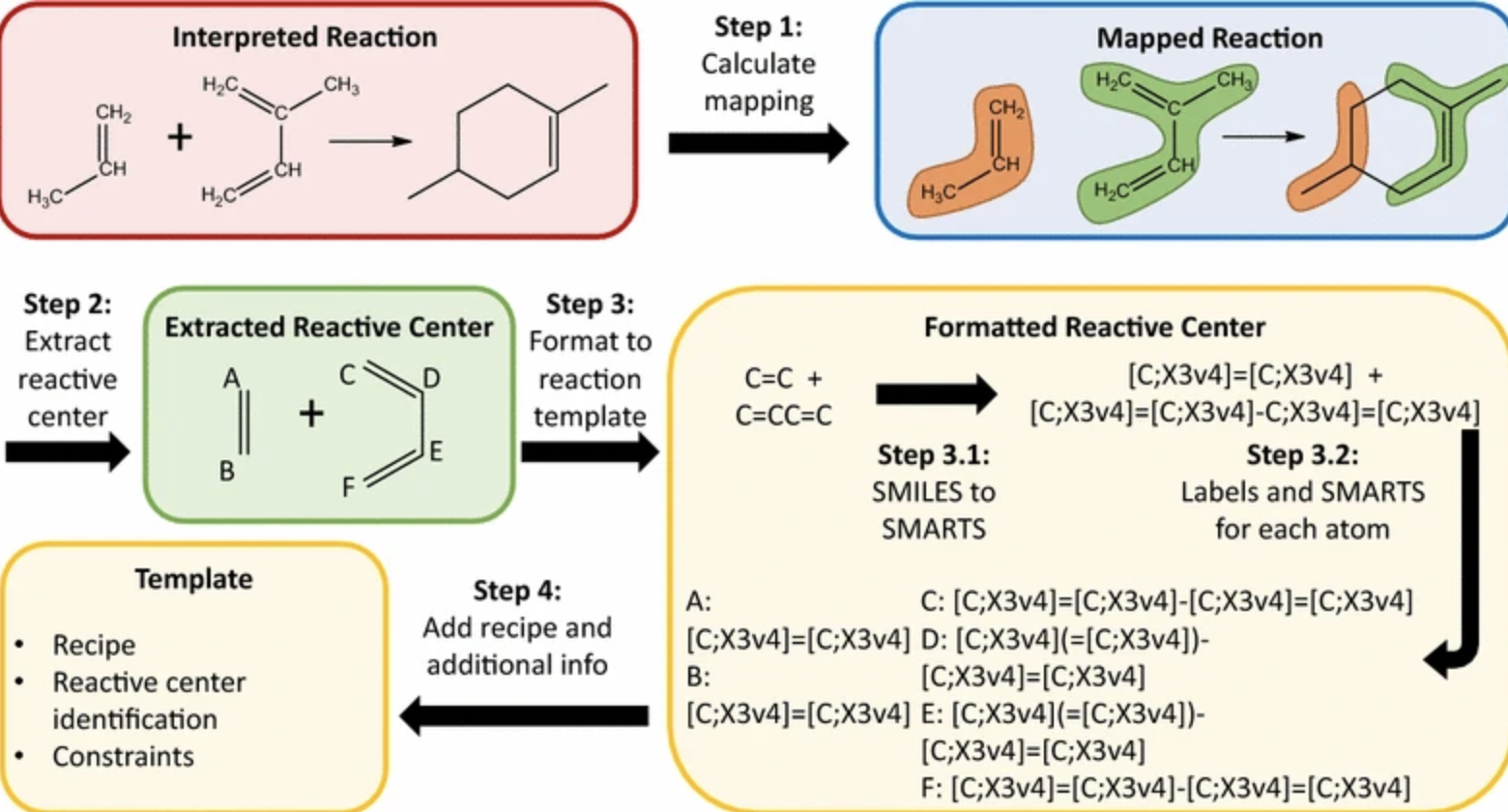 Example of reaction core extraction and reaction representation strategy, from Plehiers et al
Example of reaction core extraction and reaction representation strategy, from Plehiers et al
Challenge 2: Combinatorial explosion of biocatalytic retrosynthesis pathways
Usually, the setup for designing a restrosynthesis starts with the compound of interest, then recursively uses a set of rules to decompose the compound into substrates until each decomposed substrate is listed as available (e.g. is something you can purchase or is an available metabolite in whatever host organism you’re using). Obtaining viable retrosynthesis paths for a desired compound even with a limited set of reaction rules can be quickly become a challenging task due to combinatorial explosion of paths and products the further we decompose the target compound. While in early software this was mostly mitigated through user tweaked heuristics, eventually methods were developed that tackled this challenge head on. For example, the classic retrosynthesis software novoStoic uses a limited set of reactions based on known reactions and reaction rules to construct a network that can then be leveraged by standard graph routing algorithms. After network construction, edge cost schemes that weigh desired criterions and conditions that the reaction pathway should follow can be implemented to skew the routes in the network (e.g. we may want to find substrates that minimize cost or reactions that minimize labor). Another approach is to use generic, metaheuristic approximations to combinatorial search, like reinforcement learning in RetroPathRL.
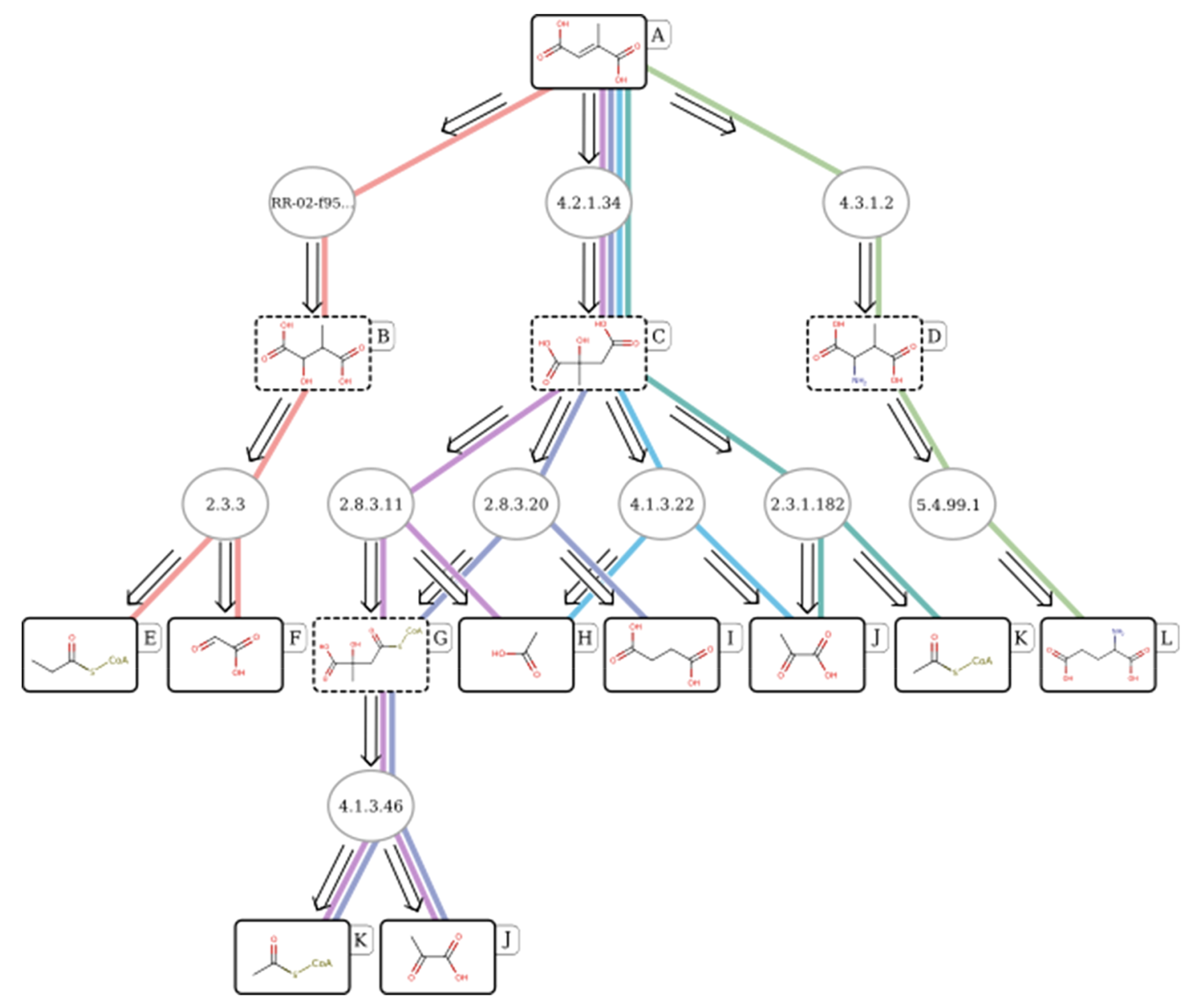 Example biocatalytic retrosynthesis pathway of mesaconic acid found by RetroPathRL, from Koch et al.
Example biocatalytic retrosynthesis pathway of mesaconic acid found by RetroPathRL, from Koch et al.
Challenge 3: Traversing enzyme sequence space during retrosynthesis
As mentioned above, an additional source of information that we can use is the aminoacid sequences and evolutionary properties of the enzymes available for retrosynthesis, giving us the ability to further transfer functional knowledge between known biocatalytic reactions. This property is exploited by some biocatalytic retrosynthesis tools, like the RetroPath family of methods, where an enzyme’s “reach” in chemical space (where “reach” is defined as either/or the span of substrates that it can process and the atomic specificity of its reaction in the substrate) can be in part learned through the aminoacid sequence. Further, enzymes themselves can be evolved, engineered, or mined for homologs that better accept the desired substrates. In an interesting extreme, one could use directed evolution to evolve all enzymes from a desired compound all the way back to a product, a sort of “retro” evoltuion that some confusingly call bioretrosynthesis. This adds another layer of empirical exploration that can be taken into account when starting a biocatalytic retrosynthesis campaign. Finally, depending on the host being used, one can tweak the amount of native enzymes and different metabolites present, some that the host might not even expect in nature, to change the metabolic flux that feeds the engineered pathway, either by making necessary metabolites more available or by out-competing natural substrates of the enzymes used for synthesis. This interplay between host and engineered enzymes need to be taken into account to avoid clashes or competition for essential substrates.
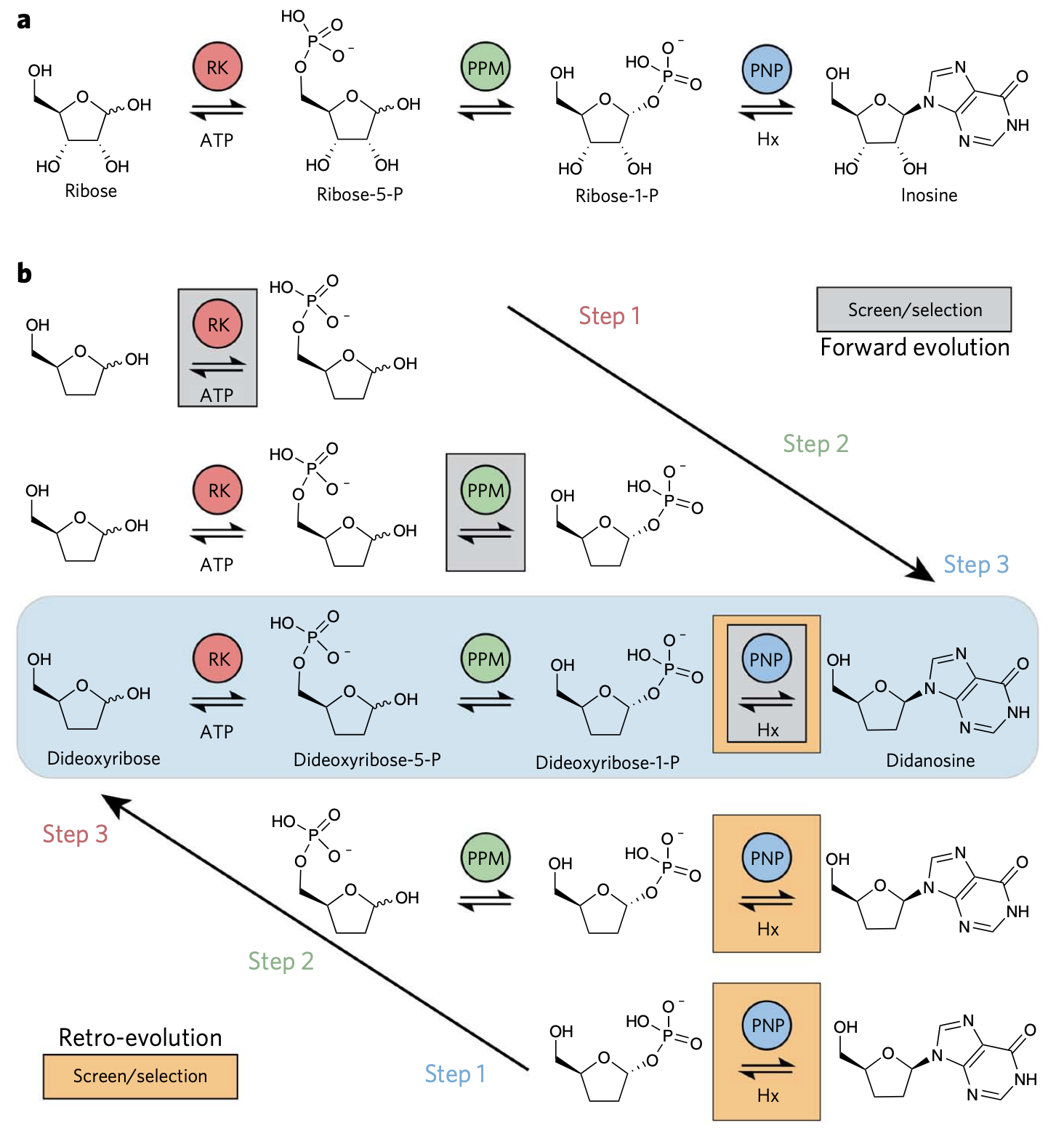 Bioretrosynthesis of inosine through forward and “retro” evolution throughout a pathway. (a) The actual inosine pathway. (b) Two strategies can be used to produce inosine, directed evolution stepwise forward from ribose, or an, in this case easier route, of evolving backward, by screening only the last step while optimizing/evolving all the enzymes required. From Birmingham et al.
Bioretrosynthesis of inosine through forward and “retro” evolution throughout a pathway. (a) The actual inosine pathway. (b) Two strategies can be used to produce inosine, directed evolution stepwise forward from ribose, or an, in this case easier route, of evolving backward, by screening only the last step while optimizing/evolving all the enzymes required. From Birmingham et al.
Challenge 4: Proper user interfacing and platform for evaluation
While the computational challenges for automating and aiding biocatalytic retrosynthesis are hard, they are not insurmountable and there already are what I thought were several good packages and software in this area. I was therefore surprised to see some reviews mention that “it’s too early to throw the towel” as if the whole effort was failing. One of my chemist colleagues mentioned that they stopped following the field some time ago and asked if it was now “ready for prime time”. Although I’m an outsider to the field, I can sort of relate to both the feelings of defeat and the skepticism. This most of the times happens where there is insufficient communication with domain experts and there is a mismatch of expectations. Perhaps the most important task of call is building good user interfaces so that domain experts can use and evaluate a tool. Recent tools like RetroBioCat fill this gap as even someone clueless like me could intuitively get it to run and was able to explore the solutions from the get-go. Proper evaluation and iteration of the tools is probably the most challenging of the tasks, as it needs either expert revision of the proposed catalytic retrosynthesis pathways or, ideally, comprehensive experimental evaluations. This last step needs laborious testing and experimental benchmarks are therefore small, on the order of testing the retrosynthesis of at most tens of compounds.
Conclusion
If all goes well, I suspect that biocatalytic retrosynthesis will be something of an emerging technology in the next few years. There is a perfect storm that has been forming for a while between increasingly complex and easy-to-deploy machine learning techniques in chemical space, maturity and commoditization of synthetic biology tools, and ubiquity of automation in biotech companies. Many of these techniques are already at the core of many companies like Zymergen and likely others will or al already following. As with many applications of computational chemistry, industry is likely the best environment to iterate and test these techniques, which has the fortunate characteristic of being quick but the unfortunate side effect that most of the advances gained remain proprietary and unpublished for a while.